In this section you will find an overview of each of the most common types of children's cancer including common symptoms, how they are diagnosed, standard treatments, new therapies being tested and current research.
There are three overall kinds of children's cancer:
Leukemia - Cancer of the Blood
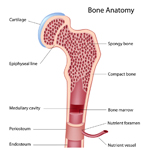
Bone marrow is a factory where our blood is made deep inside our bones. It makes red blood cells (which carry oxygen and nutrients through the body), white blood cells (which fight germs and infections) and platelets (which help stop bleeding).
Leukemia is a cancer of the blood. Leukemia cells are sick immune blood cells that do not work properly and crowd out healthy blood cells. Leukemias are the most common childhood cancers. Types of leukemia include acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML).


Lymphoma - Cancer of the Immune System
The body has a defense system, the immune system. The immune system finds cells that are not healthy or cells that do not belong in the body and destroys them. The immune system stores fighter cells, called lymphocytes, in lymphoid tissues in the body.
Lymphoma is a cancer of the immune system and lymphoid tissues. The sick cells do not work properly to protect the body and they crowd out healthy cells of the immune system. Types of lymphomas include Hodgkin disease (or Hodgkin lymphoma) and non-Hodgkin lymphoma.
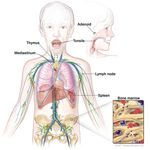

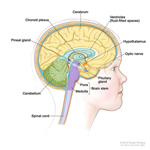
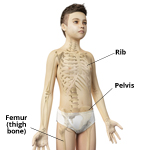
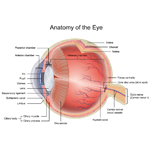
Solid Tumors - Cancer of the Bone, Organs or Tissues
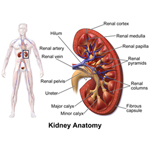
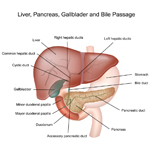
A solid tumor is a lump of sick cells stuck together. Tumors can develop in many parts of the body including the brain, kidneys, liver and bones. These sick cells crowd out healthy cells and keep them from doing their job. Types of solid tumor cancers include neuroblastoma, Ewing sarcoma and Wilms tumors.